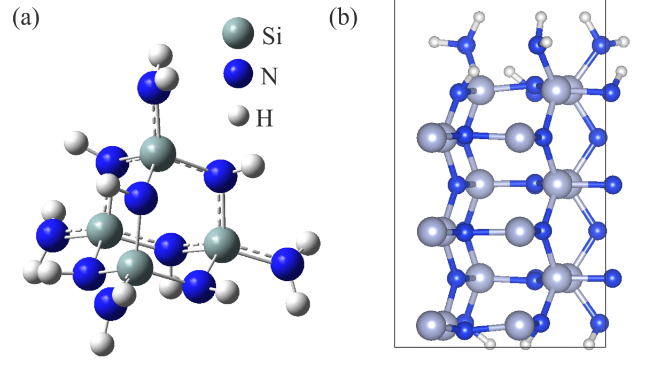
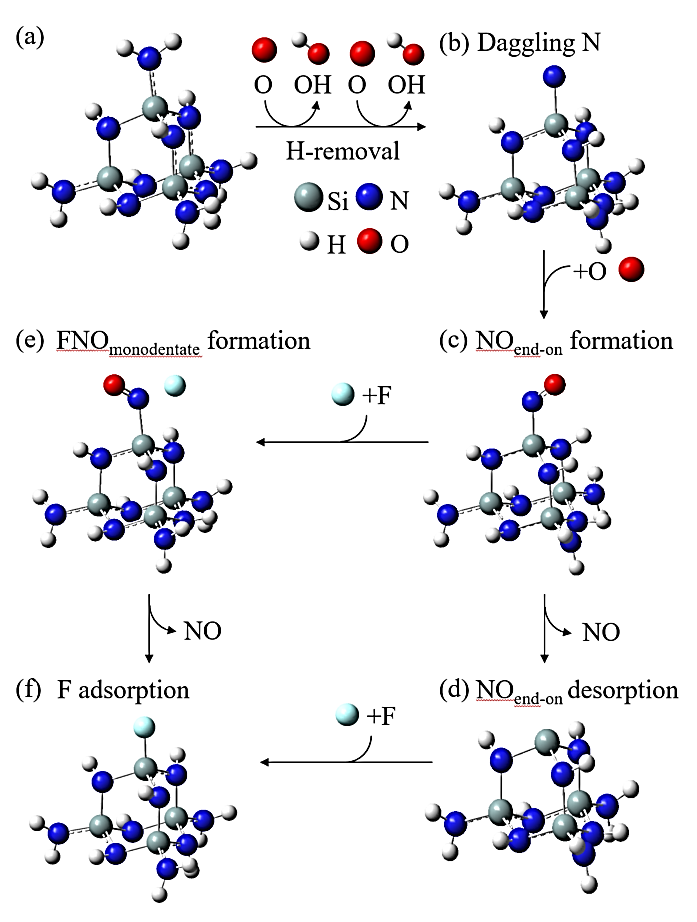
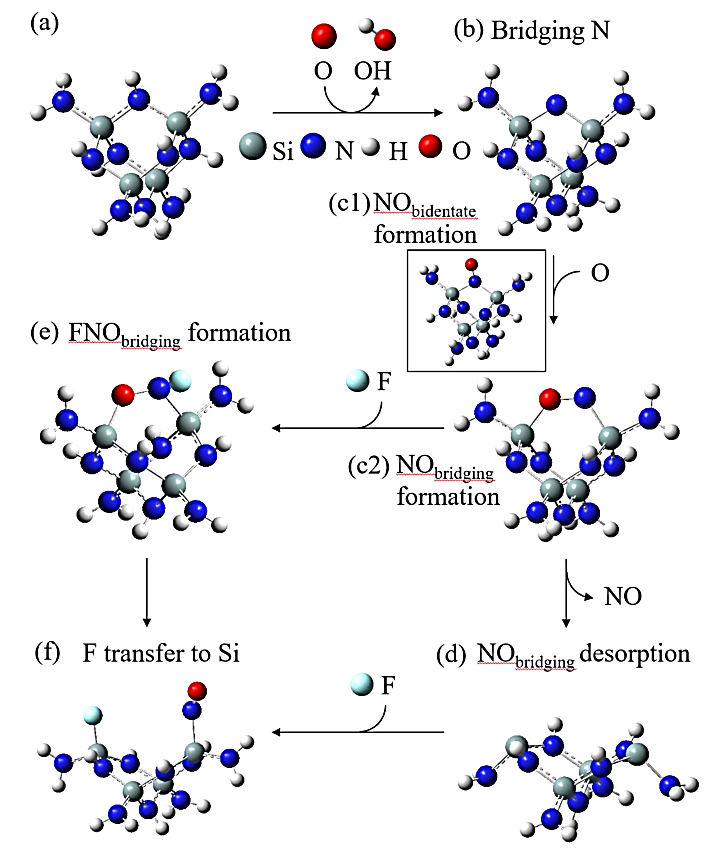
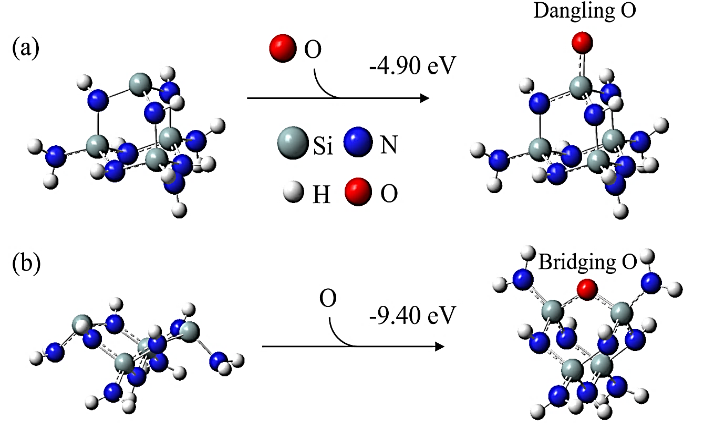
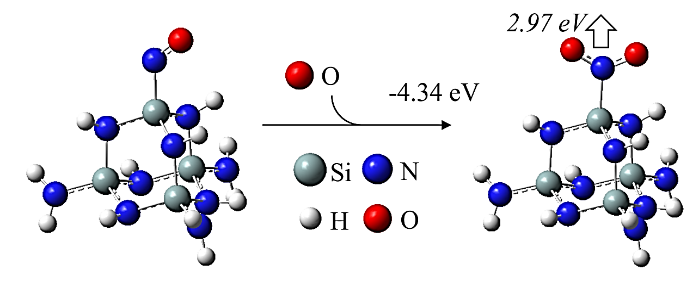
Abstract: Conducting all-in-one etch process for 3D-NAND fabrication requires close etch rate (E/R) for SiO2 and Si3N4; however, to attain comparable and high etch rate for both materials is challenging. In this work, we performed first-principle studies on the etching mechanism of Si3N4 in fluorocarbon/oxygen plasma. The feasibility of using fluorocarbon/oxygen plasma to etch Si3N4while attaining close E/R to SiO2 through the complementary nitride to oxynitiride (SiOxNy) transformation has been identified. Such transformation involves two stages: N atom elimination and Si-O bond formation. By modeling the essential chemical reactions on the Si3N4surface, we shed light upon the underlying mechanisms behind both stages. We simulated the N-elimination reactions involving the formation and desorption of NO and FNO molecules as well as the substitution with F atoms. We found that N atoms can be eliminated by forming NO molecules, especially with the assistance of F-substitution in Si-N bond breaking. The predicted O-additive energies indicates that forming SiOxNy structure after N-elimination is possible. Following that, the dependency of chemistries favoring either high E/R or active SiOxNy formation on the fluorocarbon/oxygen ratio was discussed. We hope that the work will build a foundation for future studies on pursuing all-in-one ON etch process via the surface modifications.Conducting all-in-one etch process for 3D-NAND fabrication requires close etch rate (E/R) for SiO2 and Si3N4; however, to attain comparable and high etch rate for both materials is challenging. In this work, we performed first-principle studies on the etching mechanism of Si3N4 in fluorocarbon/oxygen plasma. The feasibility of using fluorocarbon/oxygen plasma to etch Si3N4 while attaining close E/R to SiO2 through the complementary nitride to oxynitiride (SiOxNy) transformation has been identified. Such transformation involves two stages: N atom elimination and Si-O bond formation. By modeling the essential chemical reactions on the Si3N4 surface, we shed light upon the underlying mechanisms behind both stages. We simulated the N-elimination reactions involving the formation and desorption of NO and FNO molecules as well as the substitution with F atoms. We found that N atoms can be eliminated by forming NO molecules, especially with the assistance of F-substitution in Si-N bond breaking. The predicted O-additive energies indicates that forming SiOxNy structure after N-elimination is possible. Following that, the dependency of chemistries favoring either high E/R or active SiOxNy formation on the fluorocarbon/oxygen ratio was discussed. We hope that the work will build a foundation for future studies on pursuing all-in-one ON etch process via the surface modifications.
Keywords: 3D-NAND; oxide; nitride; oxynitride; plasma etch; first-principle